B. J. Sumpio1, D. Balkin4, D. Scott2, P. Le Pabic3, T. Schilling3, D. Narayan1 1Yale University School Of Medicine,Plastic And Reconstructive Surgery,New Haven, CT, USA 2Baylor College Of Medicine,Molecular And Human Genetics,Houston, TX, USA 3University Of California – Irvine,Developmental And Cell Biology,Orange, CA, USA 4University Of California – San Francisco,Plastic And Reconstructive Surgery,San Francisco, CA, USA
Introduction:
Blepharophimosis-ptosis-epicanthus inversus syndrome (BPES) is a rare disfiguring disease that results in abnormal faces. Although originally thought to be a purely soft tissue disorder, recent evidence suggests that orbital dysmorphism is also part of the disease. This includes a more lateral orbital wall, deeper orbits and flattened projections of the orbital rims. The lateral orbital wall is vertical, the orbit is deeper than normal and there is flattened projection of the orbital rims. The orbital volume can be less than normal and the supraorbital rim can be notched. The constellation of physical features are generally isolated to the periorbital region and may have some or all of the listed traits.
The physical manifestations were originally described as the result of a mutation a transcription factor gene—FOXL2 -3q23. However, 105 mutations have been associated with BPES-like phenotypes. Here we investigate a novel, previously unreported pair of genes which result in BPES when mutated.
Methods:
A male patient with BPES was identified along with the parent and siblings who had similar facial morphology. Physical features and anthropometric measurements were recorded. Whole blood samples were obtained and genomic DNA extracted. Whole exome sequencing was performed and candidate mutations identified. Sanger sequencing was performed with appropriate primers to confirm. The entire coding region of the FOXL2 gene was resequenced via the Sanger method to confirm the absence of FOXl2 mutations.
Results:
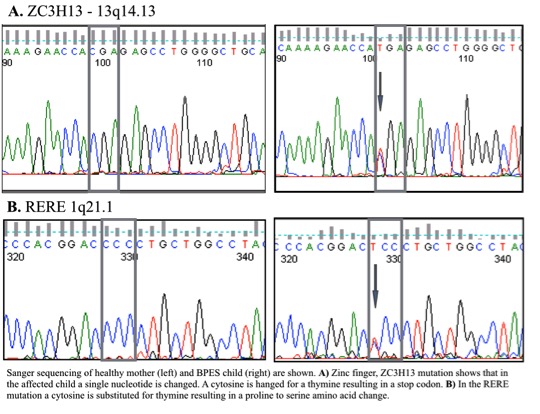
Phenotypic features of this disease were found in 2 generations of living relatives (father, 2 male children and female child) As well as documented in the grandparents as well. The inheritance demonstrated a Mendelian autosomal dominant pattern with 100% penetrance. Genetic analysis confirmed that a conserved mutation was responsible for the progression of disease, while whole exome sequencing identified candidate genes ZC3H13, and RERE with a nonsense and missense mutation, respectively.
We have identified a patient with orbitoblepharophimosis and, together with the father, the subjects were found to have a normal FOXL2 gene sequence, which was originally thought to manifest the disease. Whole exome sequencing and Sanger sequencing confirmed that FOXL2 was normal. The point mutation in ZC3H13 results in a premature stop codon of a gene which is known to be a strong transcription factor for FOXL2. Addition the single point mutation in RERE changes a cytosine for a thymine resulting in a proline to serine amino acid change.
Conclusion:
We have identified a missense and a nonsense mutation that together result in the BPES phenotype. Furthermore we have shown that FOXL2, a gene initially thought to be responsible for the mutation, to be completely normal in these patients.